泰博医疗-专业的儿科医疗连锁机构

作者: 发布时间:2015-08-14 转至微博:
|
医药网8月13日讯 有人说,“国内的产品研发方向不应该完全以抢占市场份额为核心,而应趋于围绕临床需求为导向”。笔者觉得此观点非常中肯,也符合我国国情。
且不说临床有巨大需求的小儿药物研发因成本等各种理由少人问津,各类罕见病药的研究也少有人尝试,因市场小众却成本高企,虽说国际市场预测其远景宏伟,国内还是兴趣索然。而在心血管等大病种领域,各企业扎堆上市、扎堆申报,导致CDE的审评严重塞车。
对照三版《管理办法》,从局令第37号(2002年公布,已废止)、第17号(2005年公布,已废止)、第28号(2007年开始实施)可以看出药物审评发生的变化。颠覆性的变化是仿制药和新药技术审评时限的变化,除了获得快速审评资格的新药外,37号令和17号令均规定,新药的临床研究和生产研究的技术审评时限是120日内完成,而仿制药为80日内完成;28号令则规定新药临床试验需在90日内完成,新药生产则需为150日,而改剂型和仿制药的申请需要160日。28号令第一次将五类改剂型的新药与仿制药申请并列起来,这也是考察国内企业申报实情后综合考量的结果。由于企业为适应相关规则,特别愿意在简单改剂型上花功夫,由此出现众多五花八门的剂型申报,严重增加了CDE的审评负担。
注册生态1:先“占坑”再补充
——没有仔细研究就先行申报,再陆续补充申请,成为审评重负
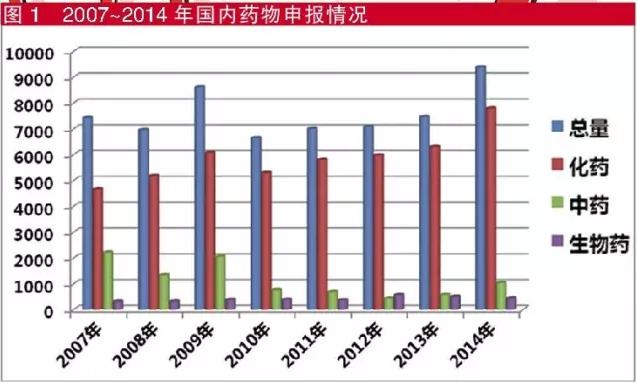
在申报数量上,虽然化药占大头的格局没有发生变化,但随着国家鼓励创新等一系列措施出台,新药的申报数量呈现快速上升的态势。然而,补充申请事项始终占据申报数量第一位的情况仍然没有改变。局令第28号令对补充申请的定义是,新药、仿制药及进口药申请批准后,如改变、增加或取消原批准事项或者内容的,均以补充申请的方式进行注册申请。这里需要说明的是,除简单的改地址等申请由各省局负责外,大多类别均需要CDE的专业技术审评。
由于药物审评采取排队进行(除快速审评),不少企业为“占坑”,在没有仔细研究的情况下以先行申报、再陆续进行补充申请的方式,达到先批准的目的。这样大大增加了审评负担,因为CDE的专业审评人员数量有限,药品的每一次补充材料,药审人员都需要从头梳理,再对材料进行评判,导致大量的时间都耗费在重复了解上。
当然,原研创新药不包括在上述情形中。现在由于不少创新药的竞争对手是国外的同行,而针对同一疾病或同一靶点的药物研发又不止一家,谁先获批谁先上临床对企业来说非常重要,所以美欧等在创新药的审批上实行的是宽进严出的政策,在安全性有保障的情况下,对其他方面暂时要求不多,之后陆续以补充申请的方式将材料补齐,这样的方式既可鼓励创新又很公平。
关键词:
|